Update: MultiQC (2019-06-21)
After I originally published this script for aggregating FASTQC reports, MultiQC was published by Phil Ewels. MultiQC aggregates quality-control and other associated data from sequencing tools into an interactive report. Instead of the script below, you can simply run:
# Run this command where your *_fastqc.zip files are
multiqc .
This will output a repor that looks like this:
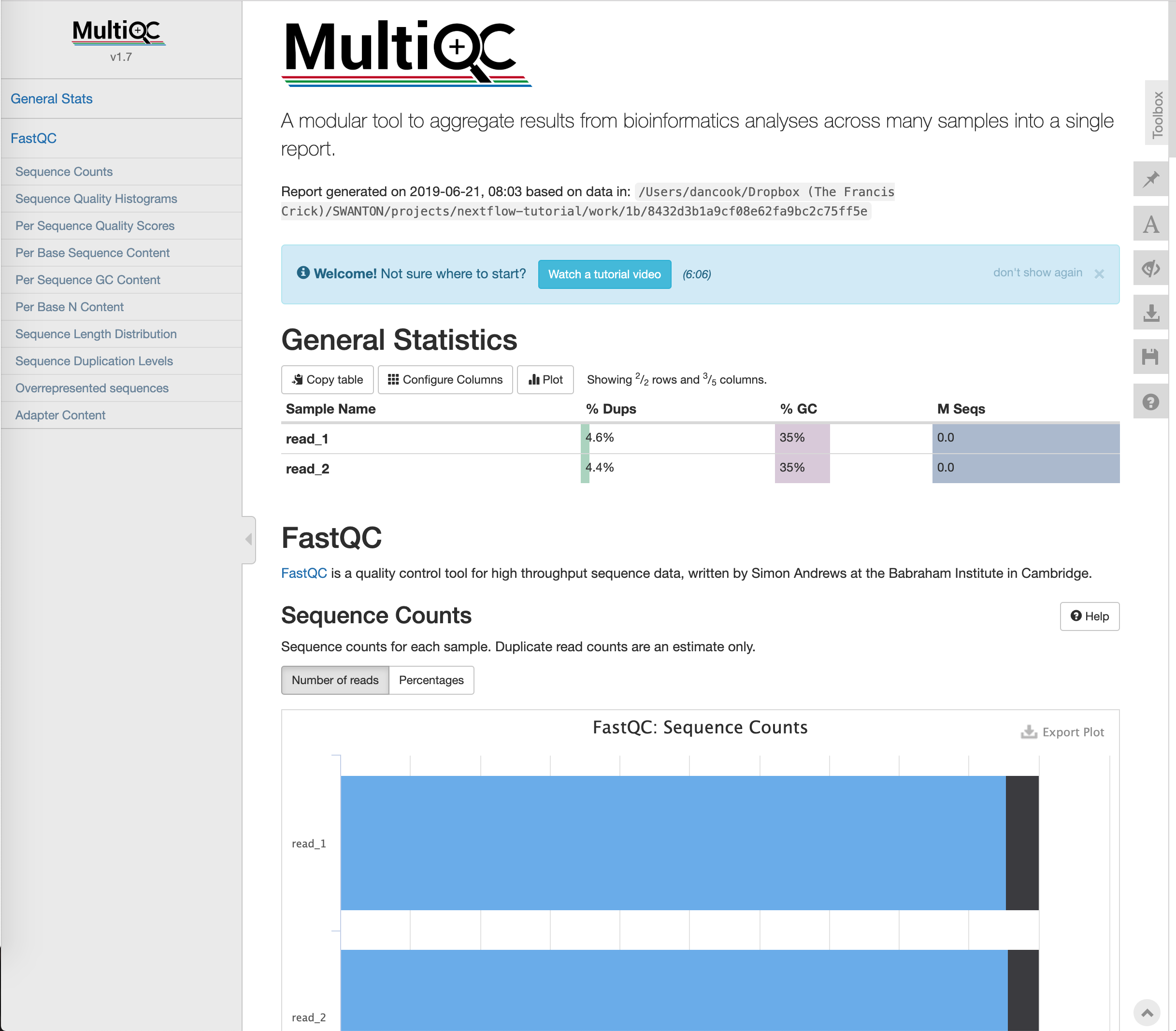
Publication
MultiQC: Summarize analysis results for multiple tools and samples in a single report Philip Ewels, Måns Magnusson, Sverker Lundin and Max Käller Bioinformatics (2016) doi: 10.1093/bioinformatics/btw354 PMID: 27312411
Original Post (2014-12-28)
FastQC is a phenomenal sequence quality assessment tool for evaluating both fastq and bam files. If you are working with a large number of sequence files (fastq), you may wish to compare results across all of them by comparing the plots that fastqc produces. I’m talking about the set of plots that look like this:
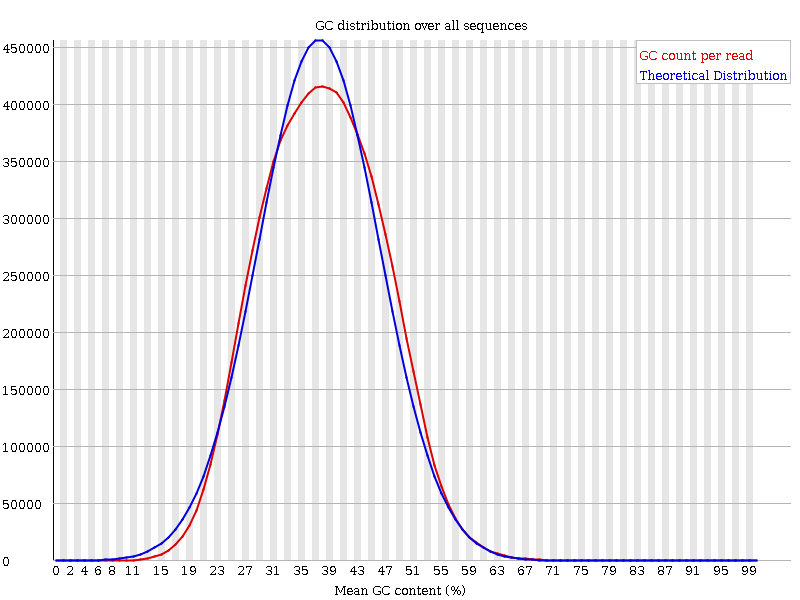
FastQC can be invoked from the command line by typing fastqc <fastq/bam>, and it will produce an html report and associated zip file containing data, plots, and some ancillary files. The zip file contains an Images folder where the plots that become incorporated into the html report are stored. They are:
- Adapter Content
- Duplication Levels
- Kmer Profiles
- Per base N Content
- Per Base Quality
- Per Base Sequence Content
- Per Sequence GC Content
- Per Sequence Quality
- Per Tile Quality
- Sequence Length Distribution
The zipped folder also contains a file called fastqc_data.txt and summary.txt. fastqc_data.txt contains the raw data and statistics while summary.txt summarizes which tests have been passed.
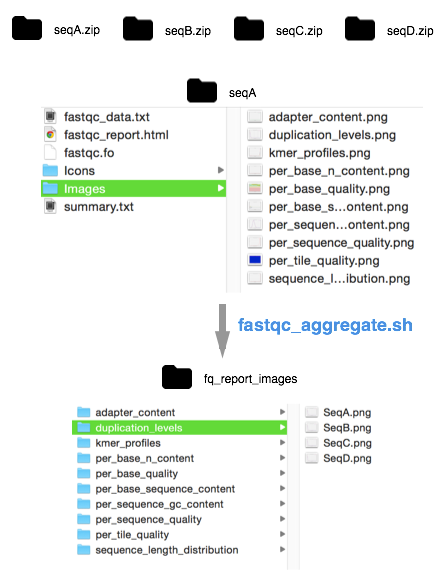
To easily compare data across reports I wrote this short shell script (below) which will ‘aggregate’ images, statistics, and summaries by:
- Unzipping all the avaible fastqc zip files.
- Creating a fq_aggregated folder, and individual folders within for each plot type.
- Move images from each unzipped fastqc report into the folder to which it belongs, and renaming it as the filename of the report (e.g. sample name).
- Concatenating summary.txt files as fq_aggregated/summary.txt.
- Concatenating the basic statistics from each report into fq_aggregated/statistics.txt.
Images will be reorganized as shown below:
summary.txt
fq_aggregated/summary.txt will produce a tab delimited file that looks like this:
| PASS | Basic Statistics | SeqA.fq |
| PASS | Per base sequence quality | SeqA.fq |
| PASS | Per tile sequence quality | SeqA.fq |
| PASS | Per sequence quality scores | SeqA.fq |
| FAIL | Per base sequence content | SeqA.fq |
| PASS | Per sequence GC content | SeqA.fq |
| PASS | Per base N content | SeqA.fq |
| … | ||
| PASS | Basic Statistics | SeqB.fq |
| PASS | Per base sequence quality | SeqB.fq |
| PASS | Per tile sequence quality | SeqB.fq |
| PASS | Per sequence quality scores | SeqB.fq |
| PASS | Per base sequence content | SeqB.fq |
| FAIL | Per sequence GC content | SeqB.fq |
| FAIL | Per base N content | SeqB.fq |
statistics.txt
fq_aggregated/statistics.txt will look like this:
| PASS | Basic Statistics | SeqA.fq |
| PASS | Per base sequence quality | SeqA.fq |
| PASS | Per tile sequence quality | SeqA.fq |
| PASS | Per sequence quality scores | SeqA.fq |
| FAIL | Per base sequence content | SeqA.fq |
| PASS | Per sequence GC content | SeqA.fq |
| PASS | Per base N content | SeqA.fq |
| d | … | |
| PASS | Basic Statistics | SeqB.fq |
| PASS | Per base sequence quality | SeqB.fq |
| PASS | Per tile sequence quality | SeqB.fq |
| PASS | Per sequence quality scores | SeqB.fq |
| PASS | Per base sequence content | SeqB.fq |
| FAIL | Per sequence GC content | SeqB.fq |
| FAIL | Per base N content | SeqB.fq |
The Code
# Run this script in a directory containing zip files from fastqc. It aggregates images of each type in individual folders
# So looking across data is quick.
zips=`ls *.zip`
for i in $zips; do
unzip -o $i &>/dev/null;
done
fastq_folders=${zips/.zip/}
rm -rf fq_aggregated # Remove aggregate folder if present
mkdir fq_aggregated
# Rename Files within each using folder name.
for folder in $fastq_folders; do
folder=${folder%.*}
img_files=`ls ${folder}/Images/*png`;
for img in $img_files; do
img_name=$(basename "$img");
img_name=${img_name%.*}
new_name=${folder};
mkdir -p fq_aggregated/${img_name};
mv $img fq_aggregated/${img_name}/${folder/_fastqc/}.png;
done;
done;
# Concatenate Summaries
for folder in $fastq_folders; do
folder=${folder%.*}
cat ${folder}/summary.txt >> fq_aggregated/summary.txt
done;
# Concatenate Statistics
for folder in $fastq_folders; do
folder=${folder%.*}
head -n 10 ${folder}/fastqc_data.txt | tail -n 7 | awk -v f=${folder/_fastqc/} '{ print $0 "\t" f }' >> fq_aggregated/statistics.txt
rm -rf ${folder}
done