Picard tools is a great set of utilities by the Broad Institute for performing sequence analysis. however, some of the utilities run on the slower side.
To speed things up, I created a new command: insert-size as part of seq-collection. The command runs much faster, owing in part to parallelization of insert-size calculations.
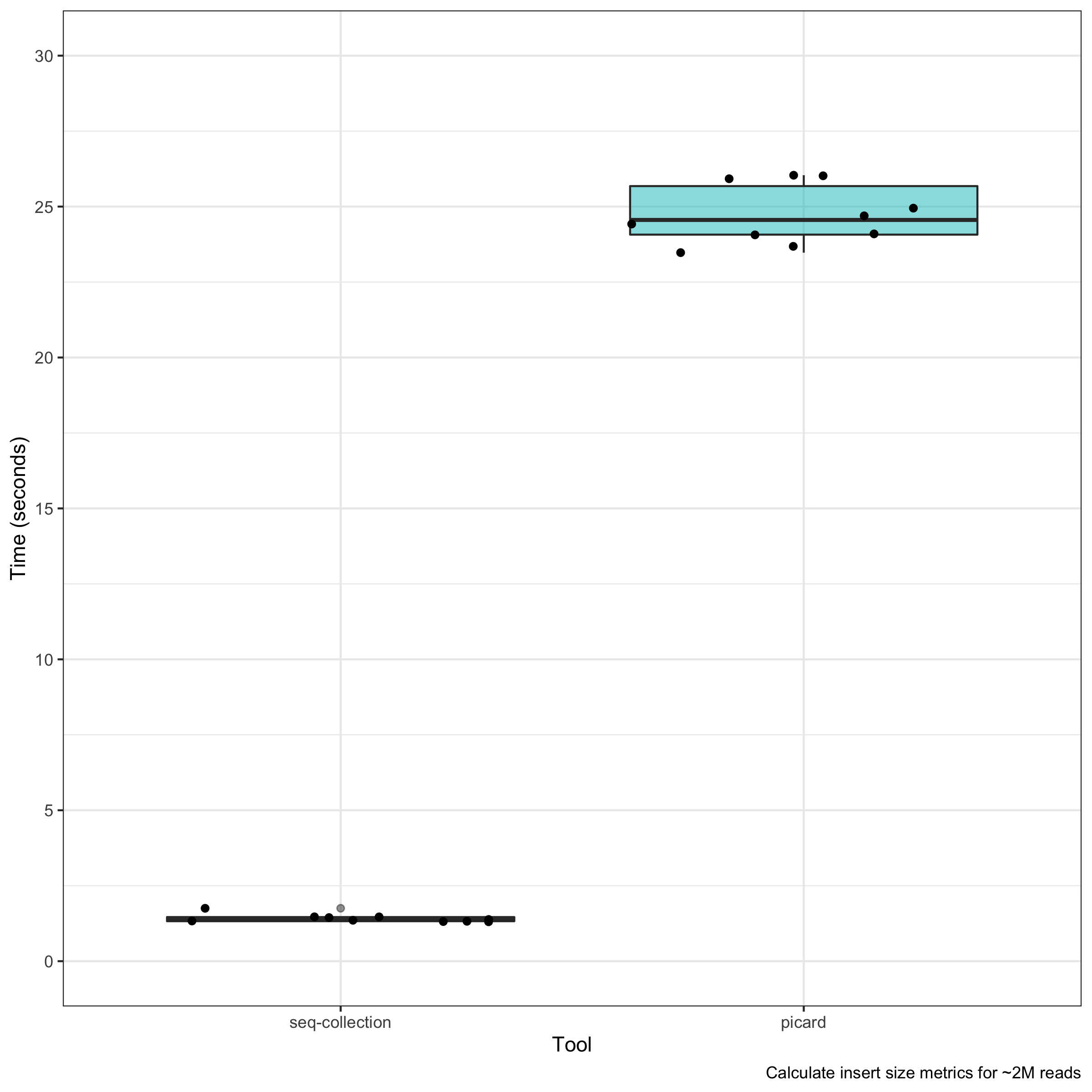
insert-size does not operate in exactly the same way as picard CollectInsertSizeMetrics, but the results are very close.
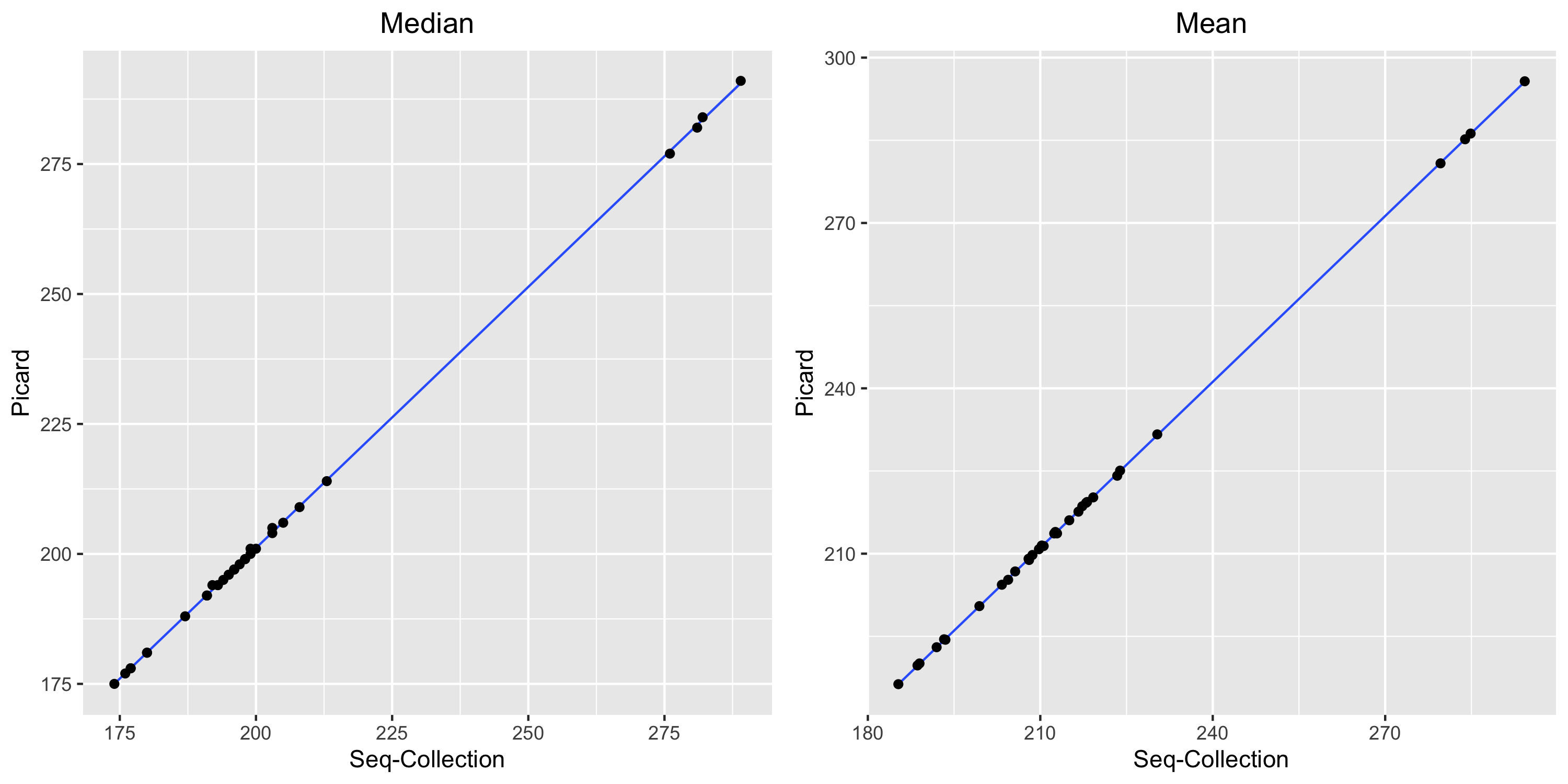
insert-size has some nice advantages over picard. The output is a lot more interpretable and parsable than standard picard output.
For example, if you run:
sc insert-size --basename --header tests/data/test.bam
The outputted table will be:
| median | mean | std_dev | min | percentile_99.5 | max_all | n_reads | n_accept | n_use | sample | basename |
|---|---|---|---|---|---|---|---|---|---|---|
| 179 | 176.5 | 63.954 | 38 | 358 | 359 | 237 | 101 | 100 | AB1 | test.bam |
You can also output the distribution of insert-sizes by count by specifying the --dist=<filename> argument.
seq-collection (sc) is a set of tools written in nim and using the fantastic hts-nim package.