If you need to annotate biological data there are plenty of resources online (UCSC Genome Browser, BioMart), and plenty of programmatic tools to interact with these databases as well. But if you are going to be annotating a large dataset (like ChIP-Seq or RNA-Seq data) – you will probably not want to rely on web based services because a) It is inefficient b) You may get throttled or banned.
If you use python, it is easy to download and store data in an SQlite database. This allows you to query the database using SQL and quickly and efficiently annotate large datasets.
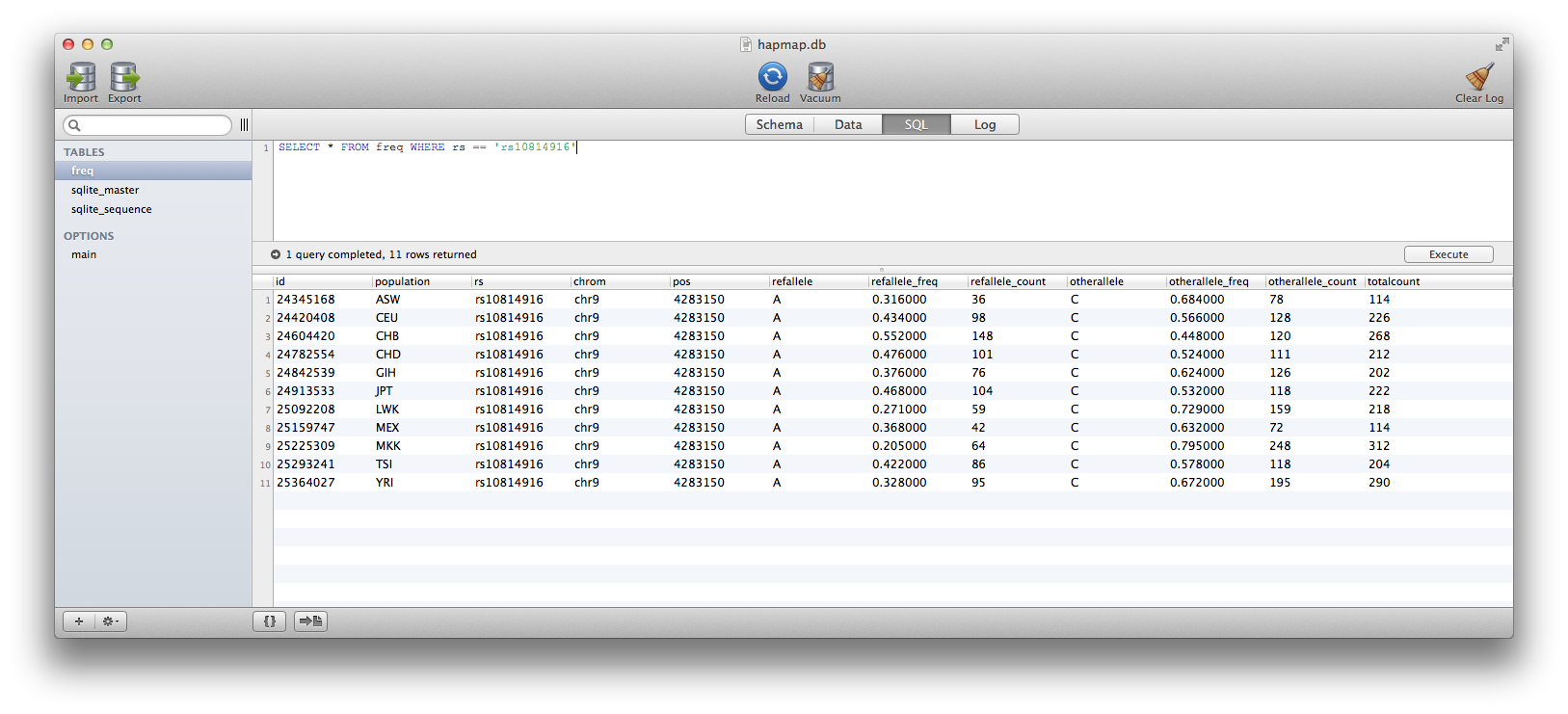
Below you will see that is what I have done here for HapMap allele frequency data (2010-08_phaseII+III), and it allows me to retrieve allele frequency data from 26,278,275 rows across 11 populations instantly. The database itself is 3.22 Gb. A zipped version (~1Gb) is available Here.
{kind=link}
You will need sqlalchemy for this script to work. Install using pip install sqlalchemy.
#! /usr/local/bin/Python
import sqlite3
import os
import glob
import time
import sqlalchemy
from sqlalchemy import Table, Column, Index, Integer, String, Float, MetaData, ForeignKey
from sqlalchemy import create_engine
import datetime
os.chdir(os.path.dirname(__file__))
os.system('wget -nd -r -A "allele*.gz" -e robots=off "http://hapmap.ncbi.nlm.nih.gov/downloads/frequencies/2010-08_phaseII+III/"')
os.system('gunzip *.gz # Unzip all the files')
if os.path.isfile('hapmap.db'):
os.remove('hapmap.db')
engine = create_engine('sqlite:///hapmap.db')
conn = engine.connect()
metadata = MetaData()
freq = Table('freq', metadata,
Column('id', Integer, primary_key=True),
Column('population', String(3)),
Column('rs', Integer),
Column('chrom', String(5)),
Column('pos', Integer),
Column('refallele',String(3)),
Column('refallele_freq',Float),
Column('refallele_count',Integer),
#
Column('otherallele',String(3)),
Column('otherallele_freq',Float),
Column('otherallele_count',Integer),
#
Column('totalcount',Integer),
sqlite_autoincrement=True,
)
metadata.create_all(engine)
for allele_file in glob.glob("allele*"):
f = file(allele_file,'r')
print f
print datetime.datetime.now()
pop = allele_file[allele_file.find('_',allele_file.find('chr')+1)+1:allele_file.find('_',allele_file.find('chr')+1)+4]
h = f.readline().replace('#','').replace('\n','')
inserts = []
c = 0
for line in f.readlines():
k = dict(zip(h.split(' '), line.split(' ')))
k['population'] = pop
c += 1
inserts.append(k)
if c == 1000:
conn.execute(freq.insert(),inserts)
inserts = []
c = 0
conn.execute(freq.insert(),inserts)
# Add indices
Index('population', freq.c.population).create(engine)
Index('rs', freq.c.rs).create(engine)
Index('chrom', freq.c.chrom).create(engine)
Index('pos', freq.c.pos).create(engine)